EMA Regulatory Pathways: How Europe Approves Medicines and What It Means for You
When you take a prescription or buy an over-the-counter drug in Europe, it didn’t just appear on the shelf—it went through a strict review by the European Medicines Agency, the independent body responsible for evaluating and supervising medicines across the European Union. Also known as EMA, it’s the gatekeeper for everything from cancer drugs to generic pain relievers. The EMA regulatory pathways are the official routes drugs must follow to get approved, and they’re not one-size-fits-all. Some medicines get fast-tracked because they treat life-threatening conditions. Others go through standard reviews that can take years. The process isn’t just about science—it’s about risk, need, and real-world use.
These pathways don’t just involve testing in labs. They rely on real patient data from clinical trials, post-market safety reports, and comparisons with existing treatments. For example, if a new drug treats a rare disease with no other options, the EMA can use accelerated approval under the conditional marketing authorization, a pathway that lets patients access promising medicines before all long-term data is in. But that comes with strings attached: the company must keep studying the drug and prove it works as expected. On the other end, generic drugs follow a different path—proving they’re bioequivalent to the brand-name version, not inventing something new. That’s why a generic pill might have different fillers or coatings, but still work the same way. And when a drug gets a boxed warning, the strongest safety alert the EMA or FDA can issue, it doesn’t mean the drug is banned—it means you need to know the risks before taking it.
These rules shape what’s available to you. A drug approved in the U.S. by the FDA might not be available in Europe for years—or ever—because the EMA has different standards. That’s why some patients travel for treatments or rely on special access programs. The EMA also monitors side effects after a drug hits the market, which is why reports of rare reactions from patients matter. If enough people report dizziness from a generic gabapentin or liver issues with a new antibiotic, the EMA can update labels, restrict use, or pull the drug. This isn’t bureaucracy—it’s a safety net. And the system isn’t perfect: delays happen, shortages occur, and not all patients get equal access. But understanding how it works helps you ask better questions, spot red flags, and know why your doctor might choose one medicine over another.
Below, you’ll find real stories and facts from patients and doctors about how these rules play out in everyday care—from how generic drugs get trusted (or not), to why some medications carry heavy warnings, and how side effects are tracked after approval. These aren’t theory pieces. They’re about what happens when policy meets practice.
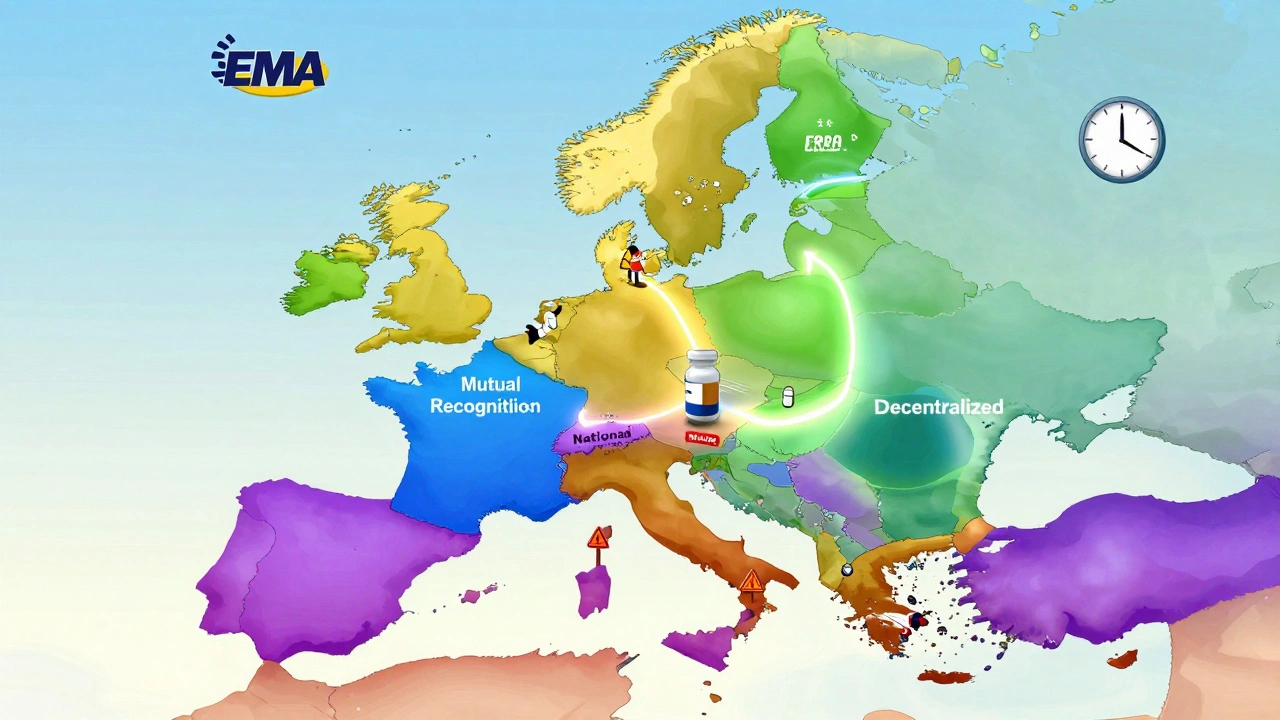
European Generic Markets: Regulatory Approaches Across the EU in 2025
The EU's generic drug market is undergoing major regulatory changes in 2025, with new approval pathways, faster market entry rules, and stricter supply obligations. Learn how the system works-and who wins and loses under the reforms.